
Introduction
Suaeda japonica Makino, belonging to the family Amaranthaceae, is an annual halophyte that grows on tidal flats in Korea, Japan, and Europe (Kim et al., 2002; Wada et al., 2003). The leaves of this plant, which is called “chilmyeoncho” in Korean, change from green to red as the plant grows. Young leaves are typically consumed as seasoned vegetables, salads, or herbs in Korea (Choi et al., 2009). S. japonica has recently been shown to exhibit a strong antioxidative activity (Choi et al., 2009; Kang et al., 2017), prevent insulin resistance (Cho et al., 2014), and inhibit tyrosinase and melanin production in B16BL6 cells (Lee et al., 2011; Lee et al., 2012). Additionally, this plant attenuates neuroinflammation in lipopolysaccharide-treated BV-2 microglial cells (Kang et al., 2013). The strong antioxidative nature of S. japonica can be attributed to its chemical constituents, including betacyanins (red pigment), betaine, 2'-hydroxy-6,7-methylenedioxyisoflavone, loliolide, and dehydrovomifoliol (Hayakawa and Agarie, 2010; Kim et al., 2005), and may help inhibit lipid peroxidation and prevent cancer and cardiovascular diseases. However, information regarding the chemical constituents of S. japonica is currently limited. In our previous study, we found 18 antioxidative compounds including flavonoids, in the aerial parts of S. japonica (Cho et al., 2013b) and identified 16 new antioxidative compounds, including coumarin glucosides, coniferyl alcohol glucosides, lignan glucosides, feruloyltyramine glucosides, and flavonol glycosides. Herein, we describe the isolation and structural determination of 16 antioxidative compounds from the aerial parts of S. japonica and evaluated the antioxidative acitivities of the isolated compounds using 1,1-diphenyl-2- picrylhydrazyl (DPPH) free radical-scavenging and cell- based reactive oxygen species (ROS) production assays.
Materials and methods
The aerial parts of S. japonica were collected from Shinan County, Korea in August 2017. The aerial parts were washed with water, dried at 50°C for 2 days using a hot air dryer (SY-8000, Samyang, Gimpo, Korea), and then ground to a powder. The powdered aerial parts were preserved at −20°C until fruther use. Hydrogen peroxide (H2O2), 2',7'- dichlorofluorescein diacetate (H2DCFDA), DPPH, quercetin, and ferulic acid were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Human hepatocellular carcinoma Huh7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Dulbecco’s modified Eagle’s medium/high glucose (DMEM) was purchased from GE Healthcare (Logan, UT, USA). Fetal bovine serum (FBS), penicillin/streptomycin (PS), phosphate buffer saline (PBS), and Hank’s balanced salt solution (HBSS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
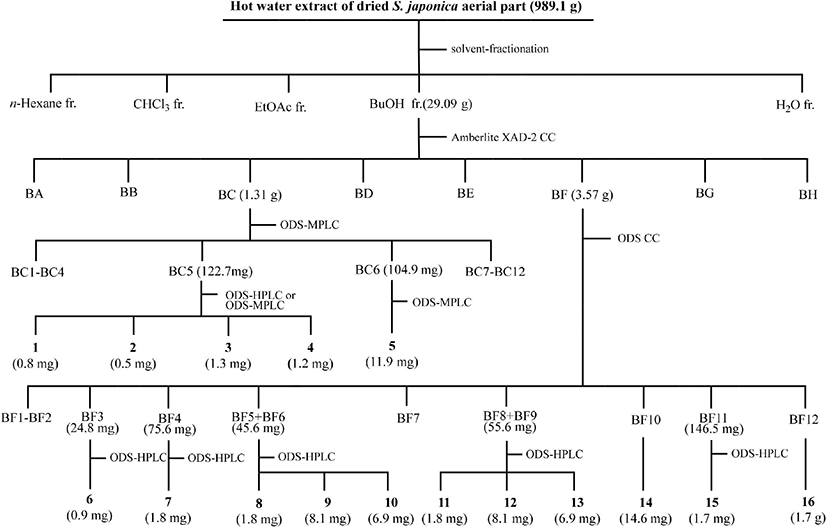
The powdered aerial parts (989.1 g) were extracted with hot water (18 L) at 90°C for 15 min and filtered through a cotton gauze. The residue was re-extracted using hot water (12 L). The hot water extract solutions were combined and partitioned with n-hexane (3×30 L), chloroform (CHCl3; 3×30 L), ethyl acetate (EtOAc; 3×30 L), and water-saturated n-butanol (BuOH; 3×30 L), successively. Each fraction was concentrated at 40°C under vacuum using an evaporator (Eyela, N-1100; Eleya, Tokyo, Japan).
The BuOH fraction (27.09 g) was loaded onto an Amberlite XAD-2 column (396.3 g, 4.0×70 cm, 587 μm; Supelco, Bellefonte, PA, USA), and eluted with a step-wise system of water (H2O)/methanol (MeOH) = 10:0, 8:2, 6:4, 4:6, 2:8, and 0:10 (v/v, 1,500 mL each step). Each fraction was spotted on silica gel thin layer chromatography (TLC) plates (silica gel 60 F 254, 0.25 mm thickness; Merck, Darmstadt, Germany) and developed with n-butanol/H2O/acetic acid = 4:1:1 (v/v). The free radical-scavenging assay was performed by spraying 200 μM DPPH radical ethanol solution onto the TLC plate. Eight fractions (BA to BH) with free radical-scavenging activities were grouped based on the silica gel TLC results. Sixteen compounds were purified and isolated from fractions BC and BF and their subfractions by medium pressure liquid chromatography (MPLC; Isolera One; Biotage, Uppsala, Sweden) coupled with a SNAP Ultra C18 column (30 g; Biotage, Sweden) and a photodiode array detector, and high performance liquid chromatography (HPLC; Shimadzu, Tokyo, Japan) equipped with a Capcell Pak C18 AQ column (4.6×250 mm, 5 μm, Shiseido, Tokyo, Japan; flow rate: 4 mL/min; UV detection: 254 nm). Fraction BC (H2O/MeOH = 8:2 [v/v], 1.31 g) was separated by octadecylsilane (ODS)-MPLC (linear gradient elution, initial H2O/acetonitrile [MeCN] = 10:0 [v/v] for 6 min → H2O/MeCN = 7:3 [v/v] for 44 min; flow rate: 25 mL/min; detection: 220 nm) to obtain 12 subfractions (BC1 to BC12). Subfraction BC5 (122.7 mg) was subjected to ODS-MPLC (linear gradient elution, initial H2O/MeCN = 10:0 [v/v] for 6 min → H2O/MeCN = 7:3 [v/v] for 44 min; flow rate: 25 mL/min; UV detection: 254 nm) to yield five subfractions (BC5a to BC5d). Compound 1 (tR = 42.1 min, 0.8 mg) was separated from subfraction BC5a (1.7 mg) by ODS-HPLC (linear gradient elution, initial H2O/MeOH = 9:1 [v/v] for 5 min → H2O/MeCN = 5:5 [v/v] for 60 min). Compound 2 (tR = 71.4 min, 0.5 mg) was purified from subfraction BC5b (tR = 31.8 min, 21.3 mg) by ODS-HPLC (isocratic elution, 25% aqueous MeCN). Fraction BC5c (19.8 mg) was subjected to the ODS-HPLC (isocratic elution, 30% aqueous MeOH) to yield 3 (tR = 28.9 min, 1.3 mg) and 4 (tR = 31.8 min, 1.2 mg). Compound 5 (tR = 71.8 min, 11.9 mg) was separated from subfraction BC6 (104.9 mg) by ODS-MPLC (linear gradient elution, H2O/MeCN = 10:0 [v/v] for 6 min → H2O/MeCN = 7:3 [v/v] for 120 min). Fraction BF (3.57 g) was chromatographed on an ODS column (300 g, 3.0×60 cm, 40-63 μm; Merck, Billerica, MA, USA) and eluted with solvent mixtures of H2O/MeCN = 90:10, 85:15, 80:20, 75:25, 70:30, 65:35, 60:40 (v/v, 750 mL each step). Ten subfractions (BF1–BF9, BF11) were grouped, and 14 (BF10; H2O/MeCN = 75:25 [v/v], 14.6 mg) and 16 (BF12; H2O/MeCN = 70:30 [v/v], 1.7 g) were purified and isolated. Compound 6 (tR = 110 min, 0.9 mg) from subfraction BF3 (isocratic elution, H2O/MeCN = 88:12 [v/v], 24.8 mg), 7 (tR = 162 min, 1.8 mg) from subfraction BF4 (isocratic elution, H2O/MeCN = 88:12 [v/v], 75.6 mg), 8 (tR = 73 min, 1.8 mg), 9 (tR = 78 min, 8.1 mg), and 10 (tR = 82 min, 6.9 mg) from subfraction BF5+BC6 (isocratic elution, H2O/MeCN = 85:15 [v/v], 45.6 mg), 11 (tR = 133 min, 1.8 mg), 12 (tR = 143 min, 8.1 mg), and 13 (tR = 162 min, 6.9 mg) from subfraction BF8+BC9 (isocratic elution, H2O/MeCN = 87:13 [v/v], 55.6 mg), and 15 (tR = 60 min, 1.7 mg) from subfraction BF11 (isocratic elution, H2O/MeCN = 87:13 [v/v], 146.5 mg) were purified by ODS-HPLC. The procedure for the purification and isolation of the 16 compounds from the BuOH layer of S. japonica aerial parts is summarized in Fig. 1.
The isolated compounds were analyzed by an electrospray ionization mass spectrometry (ESI-MS; Waters, Micromass, Manchester, UK). Nuclear magnetic resonance (NMR) spectra of the isolated compounds were acquired using unityINOVA 500 and 600 spectrometers (Varian, Walnut Creek, CA, USA). The isolated compounds were dissolved in deuterated methanol (CD3OD; δH3.31, δC49.15; Merck Co., Darmstadt, Germany), which is an analytical solvent.
Compound 1: 1H-NMR (600 MHz, CD3OD) δ 6.12 (1H, d, J = 9.6 Hz, H-3), 7.75 (1H, d, J = 9.6 Hz, H-4), 6.72 (1H, s, H-5), 7.34 (1H, s, H-8), 4.75 (1H, d, J = 7.2 Hz, H-1'); ESI-MS (positive) m/z 341.1 [M+H]+.
Compound 2: 1H-NMR (600 MHz, CD3OD) δ 6.99 (1H, d, J = 1.8 Hz, H-2), 7.02 (1H, d, J = 8.4 Hz, H-5), 6.86 (1H, dd, J = 8.4, 1.8 Hz, H-6), 6.46 (1H, d, J = 15.9 Hz, H-7), 6.20 (1H, dt, J = 15.9, 6.0 Hz, H-8), 4.13 (2H, dd, J = 6.0, 1.8 Hz, H-9), 3.79 (3H, s, 3-OCH3), 4.81 (1H, d, J = 7.8 Hz, H-1'); ESI-MS (positive) m/z 346.1 [M+Na]+.
Compound 3: 1H-NMR (600 MHz, CD3OD) δ 6.14 (1H, d, J = 9.6 Hz, H-3), 7.76 (1H, d, J = 9.6 Hz, H-4), 7.27 (1H, s, H-5), 6.73 (1H, s, H-8), 3.80 (3H, s, −OCH3), 4.75 (1H, d, J = 7.8 Hz, H-1'); ESI-MS (positive) m/z 355.1 [M+H]+.
Compound 4: 1H-NMR (600 MHz, CD3OD) δ 6.26 (1H, d, J = 9.6 Hz, H-3), 7.82 (1H, d, J = 9.6 Hz, H-4), 7.01 (1H, s, H-5), 7.20 (1H, s, H-8), 4.99 (1H, d, J = 7.2 Hz, H-1'); ESI-MS (positive) m/z 343.1 [M+H]+.
Compound 5: 1H-NMR (600 MHz, CD3OD) δ 6.46 (1H, br. d, J = 1.2 Hz, H-6), 6.76 (1H, br. s, H-8), 7.91 (1H, d, J = 1.8 Hz, H-2'), 6.87 (1H, d, J = 8.4 Hz, H-5'), 7.63 (1H, dd, J = 8.4, 1.8 Hz, H-6'), 3.79 (3H, s, 3'-OCH3), 5.05 (1H, d, J = 7.2 Hz, H-1''), 4.48 (1H, d, J = 1.2 Hz, H-1'''), 1.05 (3H, d, J = 6.0 Hz, H-6'''), 5.24 (1H, d, J = 7.2 Hz, H-1''''); 13C-NMR (125 MHz, CD3OD) δ 158.18 (C-2), 135.71 (C-3), 179.65 (C-4), 162.77 (C-5), 101.11 (C-6), 164.70 (C-7), 96.06 (C-8), 159.64 (C-9), 107.63 (C-10), 122.98 (C-1'), 114.67 (C-2'), 151.14 (C-3'), 148.47 (C-4'), 116.22 (C-5'), 123.34 (C-6'), 56.92 (3'-OCH3), 104.07 (C-1''), 76.12 (C-2''), 78.23 (C-3''), 71.90 (C-4''), 77.53 (C-5''), 68.75 (C-6''), 102.67 (C-1'''), 72.21 (C-2'''), 72.40 (C-3'''), 73.93 (C-4'''), 69.91 (C-5'''), 18.03 (C-6'''), 101.65 (C-1''''), 74.88 (C-2''''), 77.98 (C-3''''), 71.45 (C-4''''), 78.57 (C-5''''), 62.63 (C-6''''); ESI-MS (positive) m/z 787.3 [M+H]+.
Compound 6: 1H-NMR (600 MHz, CD3OD) δ 4.85 (1H, d, J = 12.0 Hz, H-2), 4.30 (1H, d, J = 12.0 Hz, H-3), 5.99 (1H, d, J = 2.4 Hz, H-6), 6.35 (1H, d, J = 2.4 Hz, H-8), 6.99 (1H, d, J = 2.4 Hz, H-2'), 6.74 (1H, d, J = 8.4 Hz, H-5'), 6.85 (1H, dd, J = 8.4, 2.4 Hz, H-6'), 3.79 (3H, s, 3'-OCH3), 4.72 (1H, d, J = 7.8 Hz, H-1''), 3.35–3.70 (4H, m, H-2''–H-5''), 3.66 (1H, dd, J = 12.0, 5.4 Hz, H-6''b), 3.85 (1H, dd, J = 12.0, 1.8 Hz, H-6''a); 13C-NMR (125 MHz, CD3OD) δ 84.68 (C-2), 74.75 (C-3), 193.64 (C-4), 166.08 (C-5), 99.43 (C-6), 168.24 (C-7), 100.42 (C-8), 162.22 (C-9), 104.37 (C-10), 129.98 (C-1'), 112.52 (C-2'), 149.06 (C-3'), 148.45 (C-4'), 116.11 (C-5'), 122.25 (C-6'), 56.59 (3'-OCH3), 104.37 (C-1''), 71.34 (C-2''), 78.73 (C-3''), 62.65 (C-4''), 77.52 (C-5''), 62.71 (C-6''); ESI-MS (positive) m/z 503.1 [M+Na]+.
Compound 7: 1H- and 13C-NMR data are given in Table 1; ESI-MS (positive) m/z 605.2 [M+Na]+.
Compound 8: 1H- and 13C-NMR data are given in Table 1; ESI-MS (positive) m/z 545.2 [M+Na]+.
Compound 9: 1H- and 13C-NMR data are given in Table 2; ESI-MS (positive) m/z 498.2 [M+Na]+.
Compound 10: 1H- and 13C-NMR data are given in Table 2; ESI-MS (positive) m/z 528.2 [M+Na]+.
Compound 11: 1H-NMR (600 MHz, CD3OD) δ 6.76 (2H, s, H-2 and H-6), 6.57 (1H, d, J = 16.2 Hz, H-7), 6.28 (1H, dt, J = 16.2, 6.0 Hz, H-8), 4.08 (2H, dd, J = 6.8, 1.8 Hz, H-9), 3.86 (6H, s, 3- and 5-OCH3), 3.37 (3H, s, 4-OCH3), 4.89 (1H, d, J = 7 .2 Hz, H-1'), 3.19–3.79 (6H, m, H-2'– H-6'); 13C-NMR (125 MHz, CD3OD) δ 135.03 (C-1), 105.65 (C-2 and C-6), 154.51 (C-3 and C-5), 136.17 (C-4), 133.70 (C-7), 126.90 (C-8), 71.46 (C-9), 105.40 (C-1'), 75.86 (C-2'), 78.51 (C-3'), 74.18 (C-4'), 77.96 (C-5'), 62.70 (C-6'), 57.15 (3- and 5-OCH3), 58.31(4-OCH3); ESI-MS (positive) m/z 409.1 [M+Na]+.
Compound 12: 1H-NMR (600 MHz, CD3OD) δ 6.61 (2H, s, H-2 and H-6), 6.60 (1H, d, J = 14.4 Hz, H-7), 5.81 (1H, dt, J = 14.4, 7.8 Hz, H-8), 4.19 (2H, dd, J = 7.8, 1.8 Hz, H-9), 3.86 (6H, s, 3- and 5-OCH3), 3.38 (3H, s, 4-OCH3), 4.92 (1H, d, J = 7.8 Hz, H-1'), 3.17-3.85 (6H, m, H-2'–H-6'); ESI-MS (positive) m/z 409.2 [M+Na]+.
Compound 13: 1H- and 13C-NMR data are given in Table 2; ESI-MS (positive) m/z 476.2 [M+H]+.
Compound 14: 1H-NMR (500 MHz, CD3OD) δ 6.14 (1H, d, J = 2.5 Hz, H-6), 6.33 (1H, d, J = 2.0 Hz, H-8), 7.59 (1H, d, J = 2.0 Hz, H-2'), 6.80 (1H, d, J = 8.5 Hz, H-5'), 7.56 (1H, dd, J = 8.5, 2.0 Hz, H-6'), 5.04 (1H, d, J = 8.0 Hz, H-1''), 4.44 (1H, d, J = 1.5 Hz, H-1'''), 3.25-3.82 (10H, m, H-2''–H-6'' and H-2'''–H-5'''), 1.05 (3H, d, J = 6.0 Hz, H-6'''); ESI-MS (positive) m/z 611.2 [M+H]+.
Compound 15: 1H-NMR (500 MHz, CD3OD) δ 6.19 (1H, d, J = 2.0 Hz, H-6), 6.38 (1H, d, J = 2.0 Hz, H-8), 8.08 (2H, d, J = 8.5 Hz, H-2' and H-6'), 6.90 (2H, d, J = 9.0 Hz, H-3' and H-5'), 5.11 (1H, d, J = 7.0 Hz, H-1''), 3.25– 3.82 (10H, m, H-2''–H-6'' and H-2'''–H-5'''), 4.53 (1H, d, J = 1.5 Hz, H-1'''), 1.14 (3H, d, J = 6.5 Hz, H-6'''); ESI-MS (positive) m/z 595.2 [M+H]+.
Compound 16: 1H-NMR (500 MHz, CD3OD) δ 6.13 (1H, d, J = 2.0 Hz, H-6), 6.33 (1H, d, J = 2.0 Hz, H-8), 7.86 (1H, d, J = 2.0 Hz, H-2'), 6.83 (1H, d, J = 8.5 Hz, H-5'), 7.54 (1H, dd, J = 8.5, 2.0 Hz, H-6'), 3.87 (3H, s, 3'-OCH3), 5.15 (1H, d, J = 7.5 Hz, H-1''), 3.25–3.83 (10H, m, H-2''– H-6'' and H-2'''–H-5'''), 4.44 (1H, d, J = 1.5 Hz, H-1'''), 1.01 (3H, d, J = 6.5 Hz, H-6'''); 13C-NMR (125 MHz, CD3OD) δ 159.00 (C-2), 135.59 (C-3), 179.47 (C-4), 163.16 (C-5), 100.13 (C-6), 166.32 (C-7), 95.07 (C-8), 158.69 (C-9), 105.81 (C-10), 123.14 (C-1'), 114.69 (C-2'), 148.46 (C-3'), 150.99 (C-4'), 116.24 (C-5'), 124.12 (C-6'), 56.91 (3'-OCH3), 104.54 (C-1''), 76.05 (C-2''), 77.52 (C-3''), 71.77 (C-4''), 78.32 (C-5''), 68.66 (C-6''), 102.66 (C-1'''), 72.22 (C-2'''), 72.41 (C-3'''), 73.97 (C-4'''), 69.93 (C-5'''), 18.03 (C-6'''); ESI-MS (positive) m/z 625.2 [M+H]+.
The free radical-scavenging activities of 1-16 and ferulic acid (positive control) were evaluated using the DPPH radical (Ban et al., 2018). Solutions of the isolated compounds were prepared at a concentration of 500 μM in dimethyl sulfoxide. The DPPH radical ethanol solution (900 μL, final concentration of 100 μM) was added to the sample solution (100 μL). Each mixture was kept in the dark for 20 min, and the absorbance was measured at 517 nm. The DPPH radical-scavenging activities of the isolated compounds were calculated as the percentage decrease in the absorbance of the sample solution relative to that of the control.
Huh7 cells were cultured in a growth medium (DMEM supplemented with 10% FBS and 1% PS) at 37°C in 5% CO2incubator. Cells seeded in a 96-well microplate (7× 103/well) were treated with the solvent-fractionated fractions (0.05 and 0.1 mg/mL), isolated compounds (20 μM), and quercetin (10 μM, positive control). After incubation for 24 h, the cells were rinsed with PBS, and 15 μM H2DCFDA in HBSS was added to the culture. After 30 min, the cells were rinsed with PBS and further incubated with 1 mM H2O2in HBSS at 37°C for 30 min. ROS levels were measured using a fluorescence microplate reader (Biotek, Winooski, VT, USA) with excitation and emission wavelengths of 485 nm and 520 nm, respectively.
Statistical analyses of the antioxidative activities of the solvent-fractionated fractions and the isolated compounds were performed using the Statistical Package for Social Sciences 20.0 package program (SPSS, Chicago, IL, USA). Statistical values were calculated using one-way analysis of variance, followed by Duncan’s multiple-range test, and presented as mean±standard deviation. Results were considered statistically significant if the p-value was less than 0.05.
Results and discussion
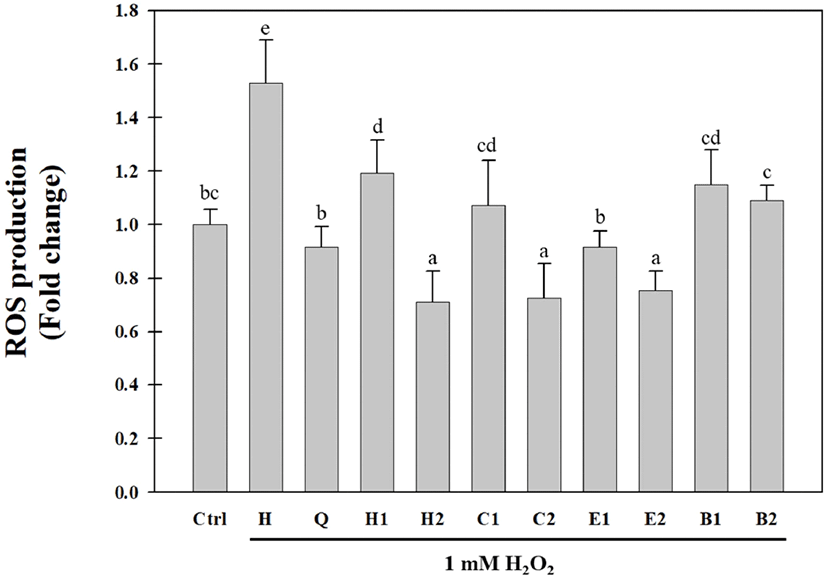
The hot water extracts of the aerial parts of S. japonica were solvent-fractionated, yielding n-hexane (0.52 g), CHCl3(0.4 g), EtOAc (1.55 g), and BuOH (27.09 g) fractions. Solvent-fractionated fractions of the aerial parts of S. japonica have been previously reported to have antioxidative activities using the DPPH radical, hydroxyl radical, hydrogen radical, hydrogen peroxide, and xanthine oxidase assays (Cho et al., 2013a; Choi et al., 2009; Kang et al., 2017). In this study, the inhibitory activities of the solvent- fractionated fractions against intracellular ROS generation in Huh7 cells were evaluated by using the H2DCFDA assay. As shown in Fig. 2, all fractions significantly decreased H2O2-induced ROS production in Huh7 cells. In particular, the EtOAc fraction (E1) at a concentration of 0.05 mg/mL exhibited the highest inhibitory activity against H2O2- induced ROS production in Huh7 cells. The ROS inhibitory activity of the EtOAc fraction (E2) at a concentration of 0.1 mg/mL was similar to those of the n-hexane (H2) and CHCl3 (C2) fractions. The BuOH fraction had a relatively low inhibitory activity against ROS generation compared to the other fractions. These results were similar to the radical-scavenging activity results in our previous study (Cho et al., 2013a; Choi et al., 2009). In addition, we identified eight antioxidative compounds, including benzoic acid derivatives, phenylpropanoids, and flavonoids from the CHCl3 and EtOAc fractions obtained after the solvent fractionation of the aerial parts of S. japonica. Compared to the n-hexane fraction, the BuOH fraction showed a low inhibitory activity against ROS production and a high DPPH radical-scavenging activity. These results suggested that various antioxidative compounds may be present in the BuOH fraction. Therefore, we attempted to purify and isolate antioxidative compounds from the BuOH fraction of the aerial parts of S. japonica.
Sixteen antioxidative compounds were purified and isolated from the BuOH fraction of the S. japonica hot water extract. Of these compounds, 11 were identified using MS and NMR data reported previously in the relevant literature. These were esculin (1) (Liu et al., 2005), citrusin D (2) (Li et al., 2011), scopolin (3) (Cho et al., 2016; Zhu et al., 2010), cichoriin (4) (EI-Bassuony and Abdel-Huamid, 2006), isorhamnetin 3-O-rutinoside-7-O-β-D-glucopyranoside (5) (Aquino et al., 1987), 3'-methoxytaxifolin 7-O-β-D- glucopyranoside (6) (Cho et al., 2013a), trans-icariside H1 (11) (Hiroyuki et al., 1991), cis-icariside H1 (12) (Masahi et al., 2005), rutin (14) (Amen et al., 2014), kaempferol 3-O-rutinoside (15) (Jung et al., 2019), and narcissin (16) (Wu et al., 2017) (Fig. 3). Two lignan glycosides (7 and 8) and three feruloyltyramine glucosides (9, 10, and 13) were structurally determined by MS and 1D- and 2D-NMR (heteronuclear single quantum coherence [HSQC], heteronuclear multiple bond correlation [HMBC], and 1H-1H correlation spectroscopy [COSY]) experiments.
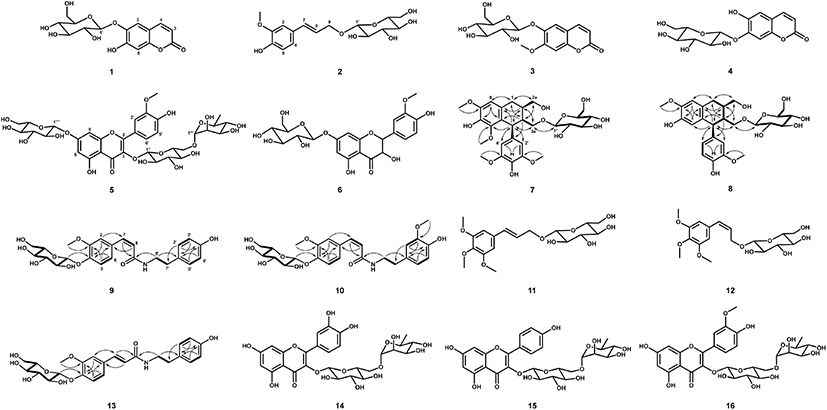
The molecular weight (522 Da) of 8 was established by observing the protonated molecular ion peak at m/z 545.2 [M+Na]+ in the ESI-MS (positive) spectrum. The 1H-NMR spectrum (500 MHz, CD3OD) of 8 showed three tri- substituted benzene ring proton signals at δ 6.79 (1H, d, J = 2.0 Hz, H-2'), 6.74 (1H, d, J = 8.0 Hz, H-5'), and 6.64 (1H, dd, J = 8.0, 2.0 Hz, H-6'), two para-benzene ring proton signals at δ 6.17 (1H, s, H-5) and 6.65 (1H, s, H-8), and two methoxy proton signals at δ 3.80 (3H, s, 7-OCH3) and 3.81 (3H, s, 3'-OCH3) (Table 1). Additionally, two methine proton signals at δ 2.07–2.12 (1H, m, H-2) and 1.83-1.88 (1H, m, H-3), one methylene proton signal at δ 2.82 (2H, dd, J = 15.0, 10.0 Hz, H-2), and three oxygenated methylene proton signals at δ 3.79–3.84 (2H, m, H-2a), 4.06 (1H, dd, J = 10.0, 2.0 Hz, H-3a), and 3.23 (1H, dd, J = 10.0, 3.0 Hz, H-3a) were observed in the 1H-NMR spectrum. The presence of the 2,3-dimethylbutan-1,4-diol moiety was confirmed by the correlations of H-1/H-2, H-2/H-2a, H-2/H-3, H-3/H-4, and H-3/H-3a in the 1H-1H COSY spectrum. The aglycone of 8 was suggested to be isolariciresinol. The presence of β-D-glucopyranose was indicated by an anomeric proton signal at δ 4.12 (1H, d, J = 7.5 Hz, H-1'') and other proton signals at δ 3.32–3.87 observed in the 1H-NMR spectrum and their coupling constant values (J = 8.0-9.0 Hz), and it was confirmed by their 1H-1H COSY correlations. The 13C-NMR spectrum of 8 showed 26 carbon signals, including eight quaternary carbonyl carbons, which were confirmed by the HSQC spectrum. The HMBC spectrum exhibited the correlations of δ 6.65 (H-8)/34.06 (C-1) and 145.95 (C-6), δ 6.17 (H-5)/48.03 (C-4) and 147.26 (C-7), δ 3.80 (7-OCH3)/ 147.26 (C-7), δ 6.79 (H-2') and 6.64 (H-6')/ 149.03 (C-3') and 48.03 (C-4), and δ 3.81 (3'-OCH3)/149.03 (C-3'), indicating that the aglycone was isolariciresinol. In particular, δ 4.12 (H-1'') was correlated to δ 69.52 (C-4) in the HMBC spectrum, indicating that C-1 of glucose was etherified to C-3a of the isolariciresinol moiety. The same configuration of H-4 and H-2 was indicated by the enhancement of H-2 at δ 2.07-2.12 from the irradiation of H-4 at δ 4.08 in the nuclear Overhauser effect (NOE) experiment. Therefore, the structure of 8 was determined to be (2R,3R,4S)-isolariciresinol 3a-O-β-D-glucopyranoside (Fig. 3).
The molecular weight of 7 (582 Da) was established by observing the sodiated molecular ion peak at m/z 605.2 [M+Na]+ in the ESI-MS (positive) spectrum. The 1H- and 13C-NMR spectra of 7 were very similar to those of 8. Compared to the 1H-NMR spectrum of 8, the spectrum of 7 showed a 2H proton signal at δ 6.41 (2H, s, H-2' and H-6') and singlet proton signal of the benzene ring at δ 6.57 (1H, s, H-8) corresponding to the galloyl moiety (Table 1). Additionally, two methoxy proton signals at δ 3.86 (3H, s, 5-OCH3) and 3.81 (6H, s, 3'- and 5'-OCH3) were observed. The 13C-NMR spectrum of 7 showed 28 carbon signals, including those of three oxygenated quaternary carbonyl carbons at δ 147.79 (C-7) and 149.25 (C-3' and C-5'). The MS and 1D-NMR results suggested that 7 was lyoniresinol glucopyranoside. Consequently, 7 was determined to be (+)- lyoniresinol 3a-O-β-D-glucopyranoside (Fig. 3) by HSQC, 1H-1H COSY, and HMBC experiments, as well as by comparing the 1D-NMR data of 7 with that of the compound isolated from Stephania hispidula (Yahagi et al., 2010).
The molecular weight (475 Da) of 9 was established by observing the sodiated molecular ion peak at m/z 498.2 [M+Na]+ in the ESI-MS (positive) spectrum. The 1H-NMR spectrum (500 MHz, CD3OD) of 9 showed the presence of a cis-ferulic acid moiety based on three tri-substituted benzene ring proton signals at δ 7.24 (1H, d, J = 2.0 Hz, H-2), 6.93 (1H, d, J = 8.0 Hz, H-5), and 6.85 (1H, dd, J = 8.0, 2.0 Hz, H-6), two olefinic double proton signals at δ 6.53 (1H, d, J = 12.5 Hz, H-7) and 5.79 (1H, d, J = 12.5 Hz, H-8), and one methoxy proton signal at δ 3.71 (3H, s, −OCH3) (Table 2). Additionally, a tyramine moiety corresponding to two p-substituted benzene ring proton signals at δ 6.57 (2H, d, J = 8.5 Hz, H-2' and H-6') and 6.87 (2H, d, J = 8.5 Hz, H-3' and H-5') and two methylene proton signals at δ 2.57 (2H, t, J = 7.0 Hz, H-7') and 3.40 (2H, t, J = 7.0 Hz, H-8') was observed in the 1H NMR spectrum. The 13C-NMR spectrum of 9 contained 16 carbon signals, including a carbonyl carbon at δ 170.14 (C-9). Based on the MS and 1D-NMR results, the aglycone of 9 was suggested to be feruloyltyramine, which is composed of ferulic acid and tyramine moieties, as identified by 1H-1H COSY, HSQC, and HMBC experiments. The 1H-1H COSY correlations of H-5/H-6, H-7/H-8, H-2' and H-6'/H-3' and H-5', and H-7'/H-8' and the important HMBC correlations of δ 6.53 (H-7) to 170.14 (C-9) and 114.67 (C-2), δ 2.57 (H-7') to 116.37 (C-2' and C-6'), and δ 3.40 (H-8') to 170.14 (C-9) established the connection of feruloyltyramine in 9 (Fig. 3). The presence of one β-D-glucopyranose was established based on an anomeric proton signal at δ 4.82 (1H, d, J = 7.5 Hz, H-1'') and other proton signals at δ 3.26-3.75 (6H, H-2''–H-6'') in the 1H-NMR spectrum and six carbon signals at δ 62.56–102.45 in the 13C-NMR spectrum, as confirmed by the 1H-1H COSY correlations. In particular, the HMBC correlation of δ 4.82 (H-1'') to 148.43 (C-4) indicated that C-1 of glucose was etherified to C-4 of feruloyltyramine (Fig. 3). Consequently, 9 was determined to be N-cis- feruloyltyramine 4-O-β-D-glucopyranoside (Fig. 3).
The 1H- and 13C-NMR spectra of 10 and 13 were similar to those of 9 (Table 2). The feruloyltyramine moieties were identical, while the methoxy group and type of olefinic double bond in these compounds differed. Compounds 10 and 13 were suggested to be feruloyltyramine glucosides. The molecular weight (505 Da) of 10 was determined from the protonated molecular ion peak at m/z 528.2 [M+H]+ in the ESI-MS (positive) spectrum. The 1H-NMR spectrum (600 MHz, CD3OD) of 10 revealed a tri-substituted benzene ring based on three proton signals at δ 6.66 (1H, d, J = 1.8 Hz, H-2'), 6.96 (1H, d, J = 9.6 Hz, H-5'), and 6.49 (1H, dd, J = 9.6, 1.8 Hz, H-6') and one methoxy proton signal at δ 3.67 (3H, s, −OCH3) (Table 2), suggesting that the partial structure of 10 was composed of dopamine (3'-methoxytyramine) instead of tyramine as in 9. The 13C-NMR spectrum of 10 showed 25 carbon signals, including one carbonyl carbon at δ 168.16 (C-9) and one methoxy carbon at δ 54.45 (-OCH3). The molecular weight (475 Da) of 13 was established by observing the sodiated molecular ion peak at m/z 476.2 [M+H]+ in the ESI-MS (positive) spectrum. The trans olefinic double bond proton signals at δ 7.36 (1H, d, J = 15.6 Hz, H-7) and 6.39 (1H, d, J = 15.6 Hz, H-8) and carbon signals at δ 141.48 (C-7) and 120.61 (C-8) were observed in the 1H- and 13C-NMR spectra of 13, and they differed from those of 9 (Table 2). Thus, compounds 10 and 13 were suggested to be N-cis-feruloylmethoxytyramine glucopyranoside and N-trans-feruloyltyramine glucopyranoside, respectively. Their accurate structures were established using 2D-NMR (1H–1H COSY, HSQC, and HMBC) spectra (Fig. 3). Therefore, 10 and 13 were N-cis-feruloylmethoxytyramine 4-O-β-D-glucopyranoside and N-trans-feruloyltyramine 4-O-β-D-glucopyranoside, respectively (Fig. 3).
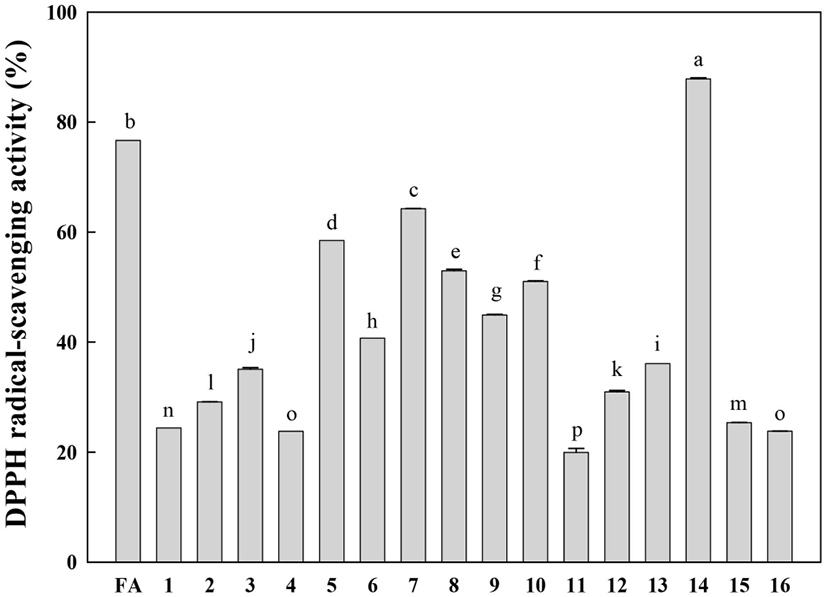
The free radical-scavenging activities of the isolated compounds (1-16) were evaluated at the same concentration (50 μM) using the DPPH radical (final concentration of 100 μM). The DPPH radical-scavenging activities of the identified compounds are shown in Fig. 4. Rutin (14) displayed the highest DPPH radical-scavenging activity. It is well known that the catechol structure is an important active site for radical scavenging by phenolic compounds (Burda and Oleszek, 2001). This study confirms that the catechol moiety is an important contributor to the excellent radical-scavenging activity of rutin (14). Compounds 5-10, which have a methoxy-substituted catechol moiety, showed relatively low DPPH radical-scavenging activities compared to rutin. Other compounds showed very low DPPH radical-scavenging activities compared to ferulic acid (positive control).
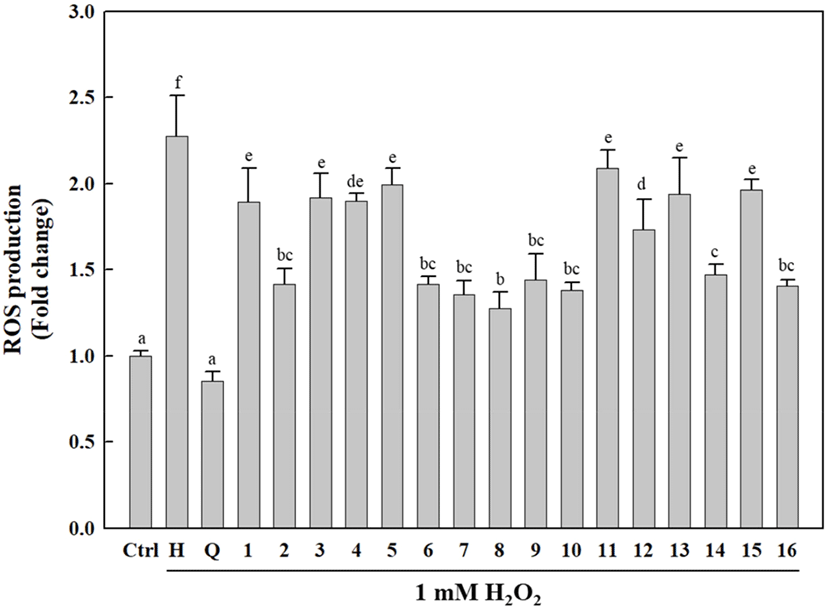
As shown in Fig. 5, the cells treated with the isolated compounds (1-16) exhibited significantly reduced intracellular ROS generation compared to H2O2-treated cells. Interestingly, the isolated compounds (1-16) differed only slightly in terms of their ROS inhibitory activities, which were relatively lower than that of quercetin (positive control). Of the isolated compounds, 2, 6-10, 14, and 16 showed higher antioxidative activities against H2O2-induced ROS generation than that induced by other compounds, with 8 exhibiting a relatively high intracellular ROS-scavenging activity compared to the other compounds. H2O2 is a toxic, strong free radical generator that induces oxidative stress during cellular metabolism (Phaniendra et al., 2015). Our results indicated that the isolated compounds (1-16) exert cellular antioxidative activities via ROS scavenging to protect cells against oxidative stress.
In this study, 16 antioxidative compounds, including three coumarin glucosides (1, 3, and 4), three coniferyl alcohol glucosides (2, 11, and 12), two lignin glucosides (7 and 8), three feruloyltyramine glucosides (9, 10, and 13), and five flavonol glycosides (5, 6, and 14-16), were isolated and identified from the aerial parts of S. japonica. To the best of our knowledge, these compounds have been identified for the first time in this plant. Among these compounds, narcissin (16) was purified and isolated in the largest amount. Narcissin has been reported to have antioxidative, antimicrobial, and immunomodulatory activities (EI-Aasr et al., 2016; Lee et al., 2003). Therefore, this compound may be used as a marker for quality control during food processing of S. japonica. In addition, the 16 antioxidative compounds identified in this study effectively scavenged the DPPH radical and reduced H2O2-induced ROS production in cells. Phenolic compounds protect cells against oxidative stress via hydrogen donation, free radical scavenging, and excessive ROS reduction (Murphy and Smith, 2007; Shirai et al., 2001; Terao, 2017). In our previous study, 16 phenolic compounds, including quercetin and luteolin, scavenged free radicals and inhibited lipid peroxidation in ferric ion- induced rat liver homogenate (Cho et al., 2013b). Thus, the various antioxidative phenolic compounds found in S. japonica can confer protection against oxidative stress and provide useful information for the utilization of S. japonica as a food and medicinal herb. Further studies on the mechanism of absorption of these antioxidative compounds in the body and the extent to which S. japonica reduces oxidative stress are necessary.