
1. Introduction
Atherosclerosis involves the build-up of lipids within the endothelium, leading to the formation of atheromatous plaques (Falk, 2006). This condition serves as the primary factor behind various cardiovascular diseases, including coronary artery disease, heart failure, stroke, ischemic gangrene, and abdominal aortic aneurysm (Hansson et al., 2006). With an increasing prevalence each year, it has become a significant global health issue, as 32% of worldwide deaths are related to cardiovascular diseases (WHO, 2017). Atherosclerosis develops through interactions of lipid-proteins, monocyte-derived macrophages, T cells, cytokines, and arterial wall cells (Glass and Witztum, 2001; Ross, 1999). The initial crucial event in atherosclerosis is the recruitment of monocytes to the arterial wall, facilitated by interactions between adhesion molecules present on the endothelium and their corresponding ligands on monocytes (Zhu et al., 2013). Pro-inflammatory cytokines and oxidized low-density lipoprotein (LDL) can increase the expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin in endothelial cells (Libby et al., 2010). The expression of vascular adhesion molecules can enhance adhesion of circulating monocytes to the endothelium and promote their transmigration into the subendothelial space (Rao et al., 2007). Tumor necrosis factor-alpha (TNF-α), a highly potent pro-inflammatory cytokine, can induce apoptosis in endothelial cells. It is also associated with the development and progression of atherosclerotic lesions (Dimmeler et al., 1999). The transcription factor nuclear factor-kappa B (NF-κB) plays an important role in regulating the expression of most inflammation-related genes (Beinke and Ley, 2004). It is particularly considered absolutely crucial for TNF-α-induced expression of VCAM and ICAM in endothelial cells (Zhou et al., 2007).
Reactive oxygen species (ROS) refers collectively to free radicals, oxygen ions, and peroxides. They can induce oxidative stress in vascular cells. They are among causes of atherosclerosis (Forstermann, 2008). Indeed, factors such as hypertension, hypercholesterolemia, diabetes, and smoking can increase ROS production in the vascular wall (Forstermann, 2008). Excessive ROS in the vasculature can enhance existing oxidative stress by inhibiting the activity of endothelial nitric oxide synthase (eNOS), leading to the generation of superoxide (Galan et al., 2014). Moreover, ROS can stimulate inflammatory activities of macrophages by inducing the expression of plaque, IL-6, and TNF-α through the NF-κB pathway (Li et al., 2014).
Angelica keiskei, a perennial herbaceous plant in the Apiaceae family, hails from the coastal areas along Japan’s Pacific coast (Amalia et al., 2021). The plant is called ‘Myeong-Il Yeob’ in Korea and ‘Ashitaba’ in Japan, both literally meaning ‘Tomorrow’s Leaf’ (Baba et al., 1998). Another common name for A. keiskei is ‘Shin-Sun Cho’, meaning ‘a precious herb used by God’ (Park, 2013). Inside the stem, there is a yellowish sap with main components including chalcone, triterpenoid, xanthoangelol, isobavachalcone, luteolin, cynaroside, and others (Kil et al., 2017). A. keiskei has been reported to possess anti-inflammatory and antioxidative activities. It has been used to treat various conditions such as diabetes, hypotension, and tumors (Jafari et al., 2023). Furthermore, A. keiskei has been reported to exhibit antithrombotic effects (Ohkura et al., 2018), improve dyslipidemia (Ogawa et al., 2005) and demonstrate preventive effects on type 2 diabetes, obesity, and atherosclerosis (Oh et al., 2019).
Currently, research specifically addressing the relationship between atherosclerosis and A. keiskei is lacking. This study aimed to bridge this gap by investigating effects of A. keiskei ethanolic extract on oxidative stress and vascular cell adhesion molecules in TNF-α-treated human umbilical vein endothelial cells. By elucidating the potential protective properties of A. keiskei in the context of atherosclerosis, this research seeks to contribute valuable insights to the existing body of knowledge.
2. Materials and methods
Quanti-MAXTM WST-8 kit and WestGlowTM FEMTO ECL Chemiluminescent Substrate were purchased from BIOMAX (Guri, Korea). Murine TNF-α was obtained from PeproTech (Cranbury, NJ, USA). 2,2'-azino-bis (3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Superoxide dismutase (SOD, cat. no. 706002), glutathione (GSH, cat. no. 703002), and catalase (cat. no.707002) assay kits were obtained from Cayman Chemical (Ann Arbor, MI, USA). Antibodies against ICAM-1, VCAM-1, E-selectin, NF-κB, p-NF-κB, IκB, p-IκB, ERK, p-ERK, p38, p-p38, JNK, pJNK, β-actin, and goat anti-mouse IgG-HRP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti-mouse IgG (H+L) Alexa Fluor plus 488 (A32723), radio-immunoprecipitation assay buffer (RIPA buffer), carboxy-H2DCFDA (general oxidative stress indicator), and eBioscienceTM Flow Cytometry Staining Buffer were obtained from Invitrogen (Carlsbad, CA, USA). Diamidino-2-phenylindole (DAPI) was came from Cell Signaling Technology (Beverly, MA, USA). Bradford’s assay reagent was came from Bio-Rad Laboratories (Hercules, CA, USA). SDS-PAGE loading buffer was purchased from Biosesang (Seongnam, Korea).
The A. keiskei used in this study was provided by 239 BIO Corporation (Iksan, Korea). The plant was identified and certified by Professor Hong Jun Kim at the College of Oriental Medicine, Woosuk University (Wanju, Korea). A reference specimen (#2023-003) has been stored in our laboratory, Department of Health Management, College of Medical Science, Jeonju University. A. keiskei was washed three times with distilled water and then cut into 1 cm sized pieces before being air-dried. The dried sample (10 g) was soaked in 200 mL of 50% ethanol and extracted at room temperature with agitation at 300 RPM for 48 h. Subsequently, the extract was filtered twice using a 0.45 μm filter paper. Ethanol was then removed using a rotary evaporator (Eyela/ N-1100S-WD, Tokyo, Japan). The remaining solution was freeze-dried to obtain a powdered form of the extract. This was named A. keiskei extract (AKE) and stored at −80°C for later use in experiments. To determine the optimal concentration of the extraction solvent, AKE was prepared using various concentrations of ethanol, and its DPPH, ABTS, and NO scavenging activities were tested. The results indicated that using 50% ethanol as the extraction solvent exhibited the highest antioxidant and anti-inflammatory effects. Therefore, in this study, AKE prepared with 50% ethanol was used.
Human umbilical vein endothelial cells (HUVECs) and Human Large Vessel Endothelial Cell Basal Medium were came from Thermo Fisher Scientific (Waltham, MA, USA). Cells were cultured at 37°C under 5% CO2 conditions.
HUVECs (2×105 cells/mL) were cultured in 96-well cell culture plate for 24 h. Subsequently, AKE (0-200 μg/mL) or TNF-α (0-60 ng/mL) was added to each well. After 20 h, Quanti-MaxTM reagent (cat. no. QM1000) (10 μL) was added to each well. Following an incubation period of 4 h, the absorbance was measured at a wavelength of 450 nm.
DPPH was dissolved in ethanol at a concentration of 0.3 mM. Subsequently, it was mixed with AKE (0-1,000 μg/mL) at a 1:1 ratio. The mixture was protected from light and allowed to react at room temperature for 20 min. Afterward, 200 μL of the solution was aliquoted into a 96-well plate, and the absorbance was measured at 540 nm using a spectrophotometer (Tecan Group Ltd., Mannedorf, Switzerland). The DPPH radical scavenging activity was calculated as a percentage using the following formula:
ABTS was dissolved in deionized water at a concentration of 7.4 mM. Potassium persulphate at 2.6 mM was then added to the solution. Radical was then generated by incubating the mixture at 4°C in the dark for 24 h. Subsequently, the solution was diluted with distilled water to achieve an absorbance of ±0.04 at 734 nm. AKE (0-1,000 μg/mL) was mixed with 50 μL of the ABTS solution. The mixture was protected from light and allowed to react at room temperature for 20 min. Following the reaction, 200 μL of the solution was aliquoted into a 96-well plate, and the absorbance was measured at 734 nm using a spectrophotometer. The ABTS radical scavenging activity was calculated as a percentage using the following formula:
HUVECs (2×105 cells/mL) were cultured in 60-mm cell culture dishes for 24 h. Subsequently, these cells were treated with AKE at concentrations of 50 and 100 μg/mL. After 1 h, TNF-α (20 ng/mL) was added. After an additional 24 h, protein extraction was performed using RIPA buffer, followed by quantification. Measurements of antioxidant enzymes were performed without modification according to the manufacturer’s protocol using the respective kits: SOD kit (cat. no. 706002), GSH kit (cat. no. 703002), and catalase kit (cat. no.707002).
HUVECs (2×105 cells/mL) were cultured in 6-well cell culture plate for 24 h. Subsequently, these cells were treated with AKE at concentrations of 50 and 100 μg/mL. After 1 h, TNF-α (20 ng/mL) was added. After 24 h, cells were collected, suspended in flow cytometry staining buffer, and incubated with Carboxy-H2DCFDA (1 μM) for 30 min. The cells were subsequently washed and examined via flow cytometry (Beckman Coulter, Brea, CA, USA) for non-fluorescent and fluorescent signals, allowing the quantification of ROS levels.
HUVECs (2×105 cells/mL) were cultured in 60-mm cell culture dishes for 24 h. Subsequently, these cells were treated with AKE at concentrations of 50 and 100 μg/mL. After 1 h, TNF-α (20 ng/mL) was added. Following protein extraction using RIPA buffer after either 30 min or 24 h, quantification was performed. Subsequently, an equal quantity of protein was separated by SDS-PAGE to segregate based on size, and then transferred onto a PVDF membrane. The membrane underwent a 1 h blocking step with 5% skim milk, followed by overnight incubation with primary antibodies VCAM-1 (1:500 dilution; cat. no. sc-13160), ICAM-1 (1:500 dilution; cat. no. sc-8439), E-selectin (1:500 dilution; cat no. sc-137054), p-NF-κB (1:500 dilution; cat no. sc-271908), NF-κB (1:500 dilution; cat no. sc-8414), p-IκB (1:500 dilution; cat no. sc-52943), IκB (1:500 dilution; cat no. sc-1643), p-ERK (1:500 dilution; cat no. sc-81492), ERK (1:500 dilution; cat no. sc-514302), p-p38 (1:500 dilution; cat no. sc-166182), p38 (1:500 dilution; cat no. sc-1972), p-JNK (1:500 dilution; cat no. sc-293136), JNK (1:500 dilution; cat no. sc-7345), and β-actin (1:1,000 dilution; cat no. sc-8432) at 4°C. Following three 10 min washes with TBST buffer, the membrane was incubated with a secondary antibody (1:5,000; sc-516102) for 2 h at room temperature. Protein expression was visualized using the WestGlowTM FEMTO Chemiluminescent substrate. ImageJ gel analysis software (National Institutes of Health, Bethesda, MD, USA) was used to analyze band density.
HUVECs (2×105 cells/mL) were cultured in cell culture slide chamber for 24 h. Subsequently, these cells were treated with AKE at concentrations of 50 and 100 μg/mL. After 1 h, TNF-α (20 ng/mL) was added. After 30 min, the culture medium was removed and cells were fixed in 4% formaldehyde. Subsequent steps included a 10 min incubation with 0.1% Triton X-100, a 1 h blocking step with 1% BSA, overnight incubation with the primary antibody (1:50 dilution; cat no. sc-271908) at 4°C, washing, and a 2 h incubation with the secondary antibody (1:5,000; cat. no. A32723) at room temperature. After completion, slides were observed using a fluorescence microscope (ZEISS, Oberkochen, Germany).
Experimental values are presented as mean±standard deviation (mean±SD). Statistical analyses were conducted using IBM SPSS Statistics 22 (IBM, New York, NY, USA). Inter-group comparisons were made using one-way analysis of variance (ANOVA), with Duncan’s multiple range test applied for post hoc analysis to identify significant differences among the groups. A significance level of p<0.05 was used for all statistical tests.
3. Results and discussion
Table 1 presents the DPPH and ABTS radical scavenging activities of AKE. DPPH and ABTS radical scavenging activities are reported as IC50 values, which represent the concentration needed for the sample to inhibit 50% of the radical activity compared to the control group, based on absorbance change curves relative to sample concentration. The DPPH radical scavenging activity IC50 value of AKE was 375.24±8.56 μg/mL, and that of ascorbic acid, a reference drug, was 23.16±3.21 μg/mL. For ABTS, AKE was measured at 568.12±9.23 μg/mL, and ascorbic acid was measured at 35.26±5.57 μg/mL. These results indicate that the ability of AKE to scavenge DPPH and ABTS radicals is less effective than that of ascorbic acid. However, AKE has potential as an antioxidant because it effectively scavenges radicals at relatively low concentrations.
| Samples | DPPH1) radical scavenging activity (IC50: μg/mL) | ABTS2) radical scavenging activity (IC50: μg/mL) |
|---|---|---|
| AKE | 375.24±8.563) | 568.12±9.23 |
| Ascorbic acid | 23.16±3.21 | 35.26±5.57 |
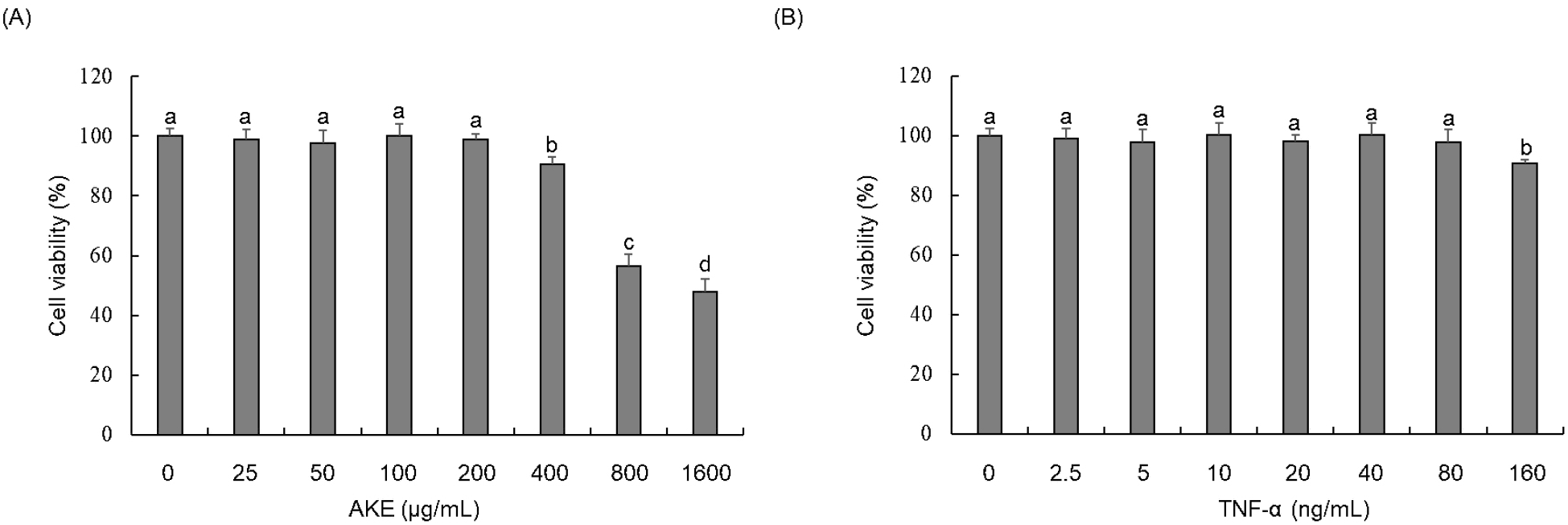
A WST assay was conducted to investigate the cytotoxicity of AKE and TNF-α in HUVECs. Results showed no toxicity for AKE up to 200 μg/mL (Fig. 1A). TNF-α showed no toxicity up to 80 ng/mL (Fig. 1B). Therefore, in subsequent experiments, AKE was administered to cells at concentrations equal to or below 200 μg/mL. TNF-α was applied at concentrations equal to or below 80 ng/mL. TNF-α did not exhibit toxicity to HUVEC at concentrations up to 80 ng/mL, but excessive amounts of TNF-α can induce non-physiological responses or saturate cell receptors, potentially distorting the results. Therefore, we decided to use a treatment concentration of 20 ng/mL, which is commonly used in studies of endothelial cell damage induced by TNF-α (Ismail et al., 2018; Zhu et al., 2013).
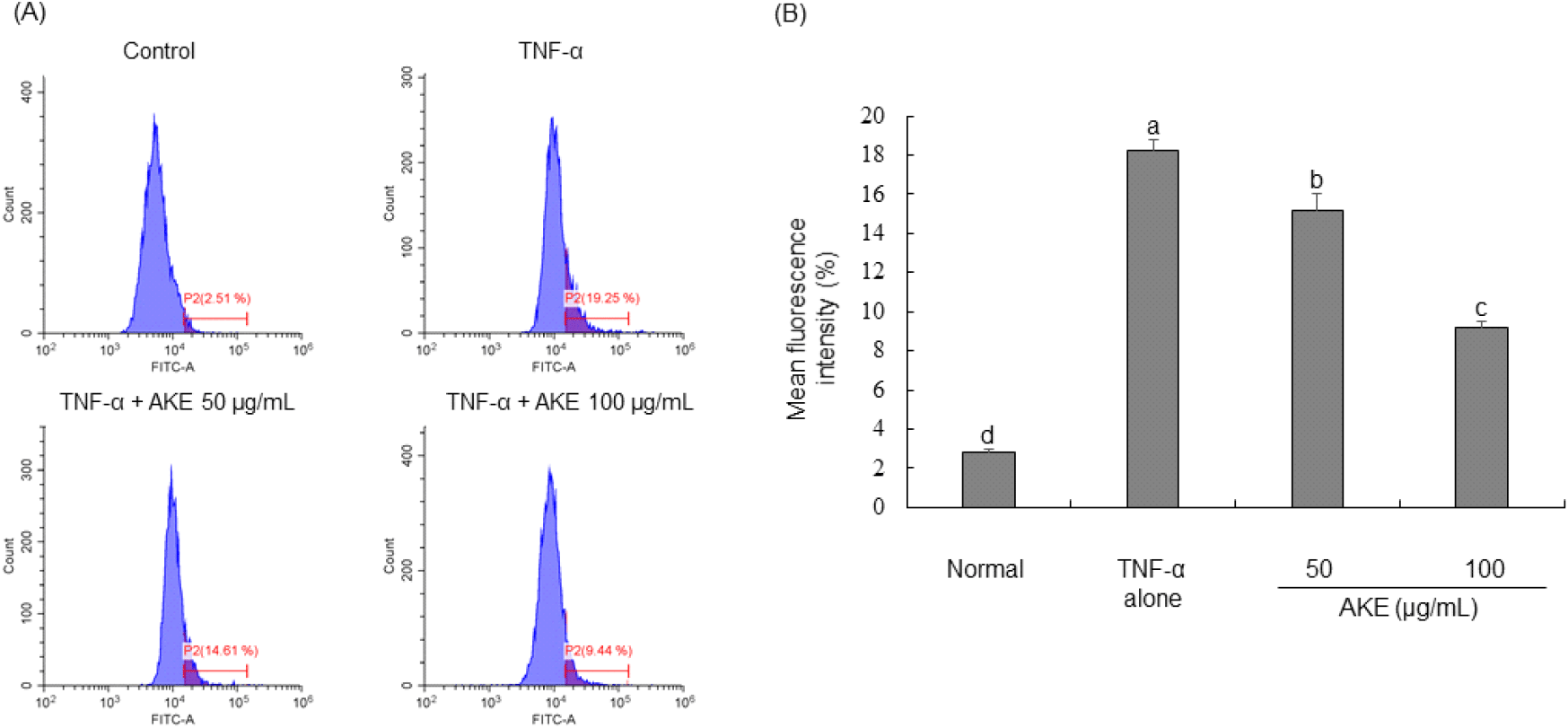
In order to investigate the impact of AKE on intracellular reactive oxygen species (ROS), carboxy-H2DCFDA was administered to TNF-α-treated HUVECs followed by flow cytometric analysis. Results demonstrated that TNF-α treatment led to a significant increase in intracellular ROS levels. In contrast, treatment with AKE dose-dependently reduced previously elevated ROS levels (Fig. 2). Additionally, treatment with AKE up to the non-toxic maximum concentration of 200 μg/mL showed no significant difference compared to 100 μg/mL (data not shown). In pharmacology, it is preferred to use the lowest effective dose of a substance to minimize side effects and toxicity. Natural products have relatively fewer side effects than drugs, but to minimize the risk of potential side effects and toxicity, AKE was tested up to 100 μg/mL, the minimum dose that shows the best effect in cells. Previous studies have documented that TNF-α stimulation can induce the generation of ROS in endothelial cells (Cominacini et al., 2002; Kim et al., 2008). Consistent with these previous findings, our study also observed a significant increase in intracellular ROS in HUVECs upon treatment with TNF-α. Under normal conditions, cells maintain a balance between ROS production and removal. However, chronic inflammation can sustainably generate high levels of ROS, leading to oxidative stress (Pan et al., 2016). AKE effectively scavenged DPPH and ABTS radicals and also reduced intracellular ROS induced by TNF-α. Therefore, we demonstrate the potential of AKE as a beneficial agent in managing conditions associated with excessive ROS production and chronic inflammation. Follow-up studies, including animal and clinical trials, are needed to determine whether the in vitro antioxidant effect of AKE can be replicated in vivo and to confirm its efficacy and safety in clinical applications.
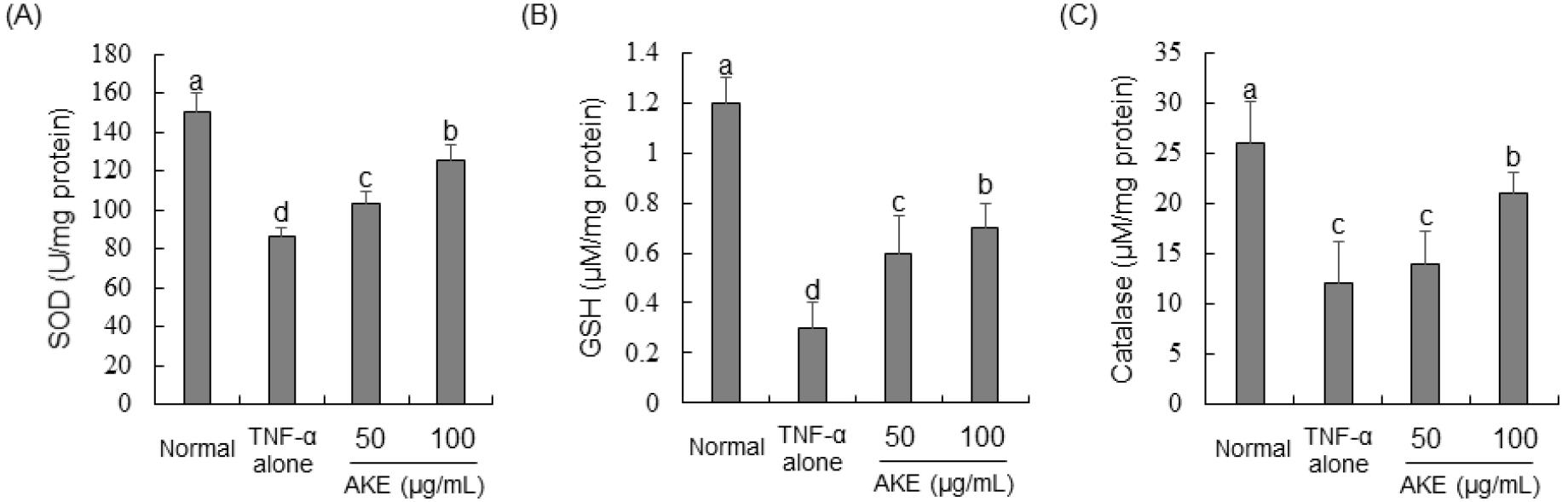
To investigate the influence of AKE on activities of antioxidant enzymes (SOD, GSH, and catalase), protein samples from TNF-α-treated HUVECs were utilized to measure the activity of each enzyme. Results revealed that TNF-α treatment significantly inhibited activities of SOD, GSH, and catalase enzymes. In contrast, activities of SOD and GSH showed concentration-dependent increases in AKE-treated (Figs. 3A and 3B). For catalase, a significant increase in its activity was not observed in cells treated with AKE 50 μg/mL. However, after treatment with AKE at 100 μg/mL, there was a noticeable enhancement in catalase activity (Fig. 3C). GSH, catalase, and SOD are vital antioxidant enzymes crucial for shielding cells against oxidative damage. They function by eliminating reactive oxygen species, breaking down hydrogen peroxide into harmless water and oxygen, and transforming superoxide radicals into oxygen and hydrogen peroxide, respectively (Ma et al., 2017). Considering this, the ROS-reducing effect of AKE shown in Fig. 2 appears to be due to increased activity of antioxidant enzymes or direct radical scavenging ability, as shown in Table 1. This suggests that AKE could restore the balance of the redox state in endothelial cells impaired by TNF-α, highlighting its potential therapeutic value in oxidative stress-related vascular diseases.
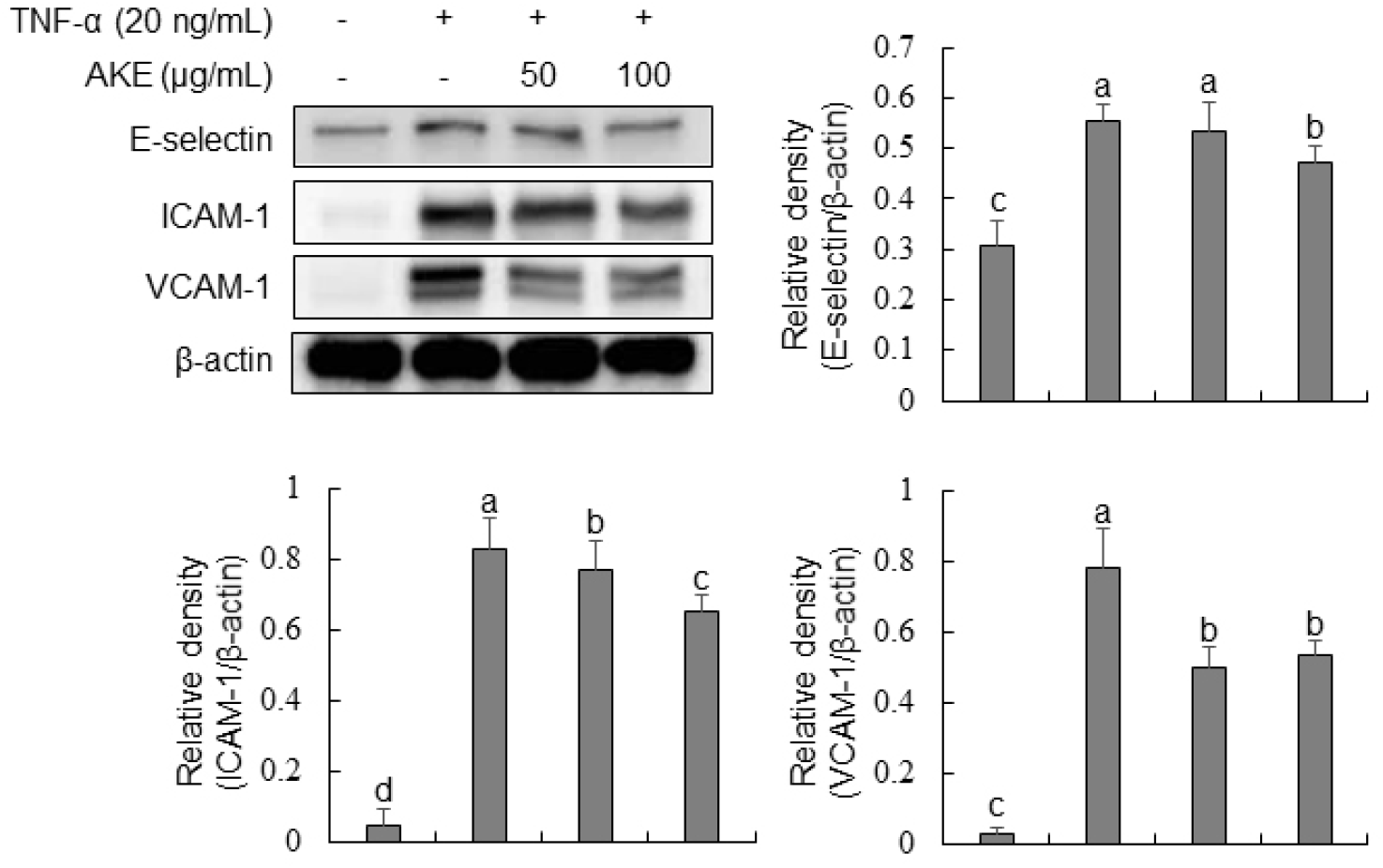
To investigate the influence of AKE on the expression of vascular adhesion molecules, Western blot analysis was conducted for TNF-α-treated HUVECs. Results demonstrated a marked rise in the expression of E-selectin, ICAM-1, and VCAM-1 in the TNF-α-treated group (Fig. 4). In the case of E-selectin, there was no change after treatment with AKE at 50 μg/mL. However, a significant decrease was observed at AKE 100 μg/mL. For ICAM-1 and VCAM-1, there was a concentration-dependent decrease in expression at AKE 50 and 100 μg/mL concentrations. TNF-α, a crucial cytokine widely utilized in the study of molecular mechanisms associated with vascular adhesion molecules in HUVECs, plays a key role in the recruitment and activation of inflammatory cells (Wu et al., 2015; Zhang et al., 2002). E-selectin, ICAM-1, and VCAM-1 play important roles in the initiation of early atherosclerosis. They are expressed during inflammatory conditions. They can mediate leukocyte movement across the endothelial barrier (Wu et al., 2015). Additionally, as a key mediator in signaling pathways, ROS can regulate the expression of vascular adhesion molecules (Cominacini et al., 2002; Kim et al., 2008; Park et al., 2010). Thus, the proven inhibitory effect of AKE on the expression of vascular adhesion molecules in this study is also presumed to be a result of ROS elimination. These findings imply that AKE may have therapeutic potential in preventing or treating inflammatory vascular diseases by reducing the endothelial expression of adhesion molecules and thereby limiting leukocyte recruitment and adhesion.
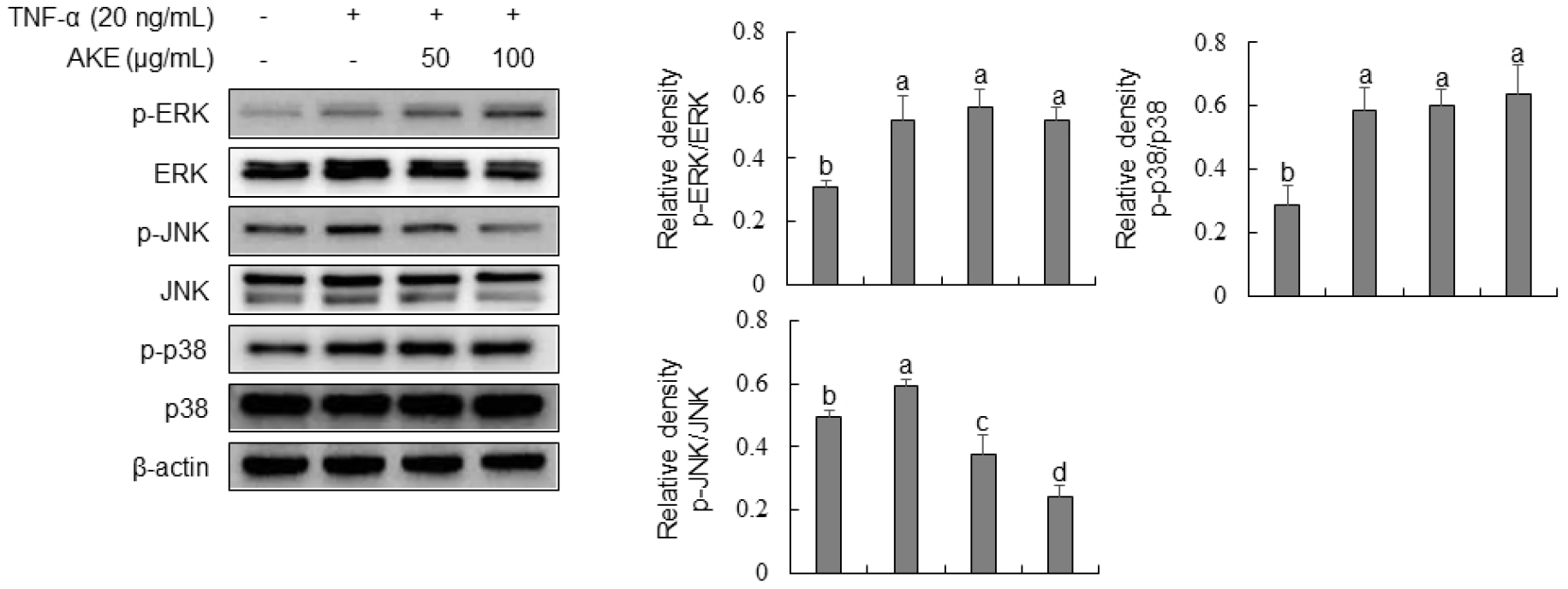
We evaluated the effect of AKE on the phosphorylation of MAPK by Western blot analysis to investigate its mechanism of action, as AKE effectively inhibited the expression of adhesion molecules in TNF-α-stimulated HUVECs. Results showed that TNF-α treatment significantly increased the phosphorylation of ERK, JNK, and p38 (Fig. 5). In contrast, AKE treatment concentration-dependently decreased the phosphorylation of JNK. ERK and p38 showed no significant changes in phosphorylation after AKE treatment. Mitogen-activated protein kinases (MAPKs), such as p38, extracellular signal-regulated kinases 1 and 2 (ERK1/2), and Jun N-terminal kinases (JNKs), are involved in a variety of signaling pathways within cells. They are known to regulate gene transcription, protein synthesis, and cell cycle control (Widmann et al., 1999). In endothelial cells, p38 and JNK are activated by TNF-α stimulation, inducing the expression of pro-inflammatory molecules (Hoefen and Berk, 2002). ERK, including ERK1/2 and BMK1, plays a crucial role in promoting cell growth and survival (Hoefen and Berk, 2002). Additionally, members of MAPKs can regulate the NF-κB-dependent pathway (Schulze-Osthoff et al., 1997). In this study, we demonstrated that AKE could inhibit the phosphorylation of JNK induced by TNF-α in HUVECs, while it did not affect the phosphorylation of ERK1/2 or p38. This finding aligns with previous reports indicating selective inhibition of JNK by compounds such as Glycyrrhetinic acid in TNF-α-stimulated HUVEC and Berberine in LPS-stimulated HUVECs (Chang et al., 2010; Guo et al., 2016). These results suggest that the anti-inflammatory effects of AKE may be primarily mediated through the inhibition of JNK signaling, rather than through ERK or p38 pathways.
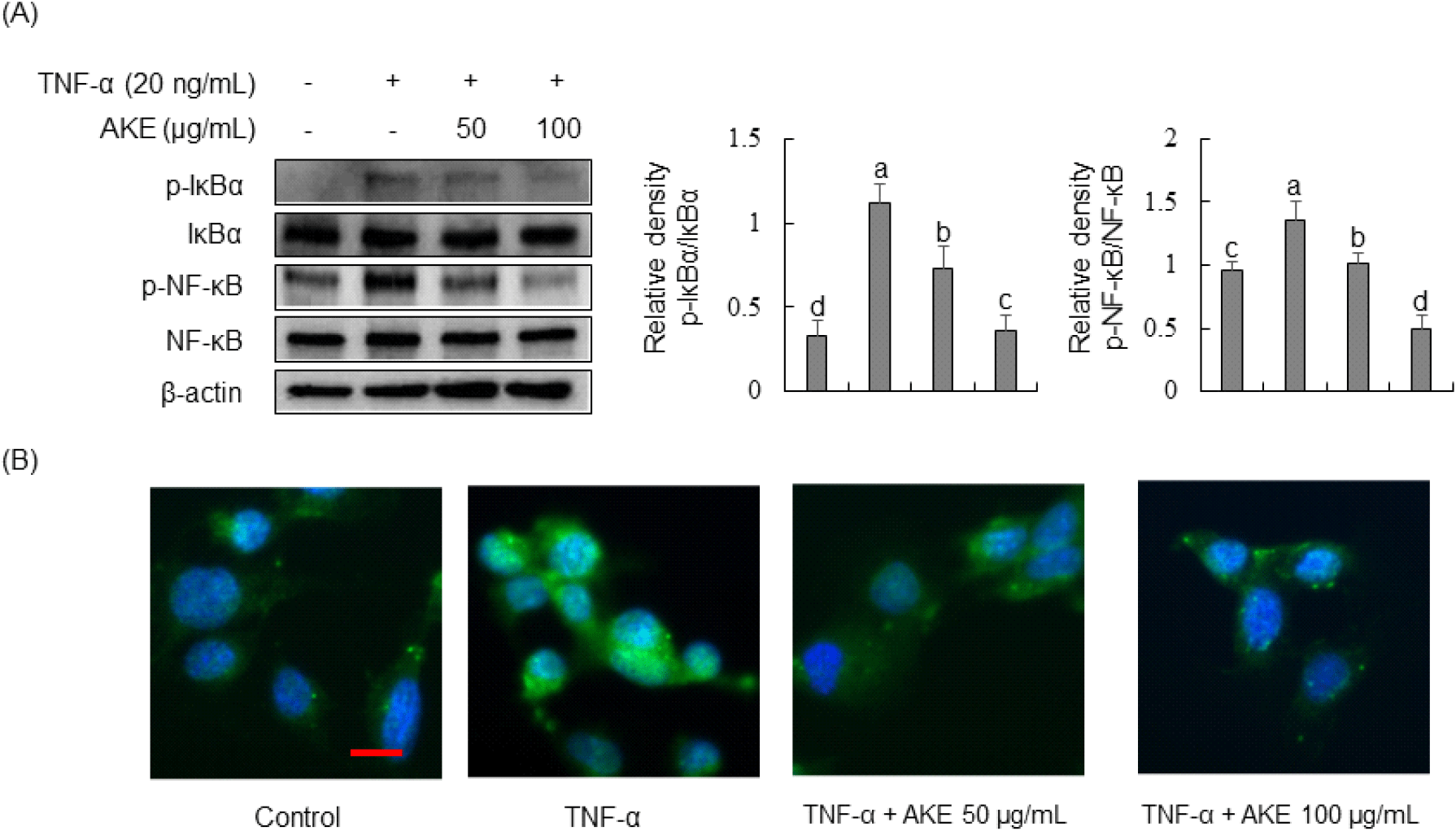
Due to inhibition of JNK phosphorylation by AKE, we investigated the impact of AKE on the NF-κB signaling pathway, a JNK cascade member. Results revealed that TNF-α treatment for 30 min significantly increased the phosphorylation of NF-κB and its inhibitor, IκBα (Fig. 6A). In contrast, AKE treatment concentration-dependently decreased the phosphorylation of IκBα and NF-κB. Immunofluorescence staining was performed to observe NF-κB nuclear translocation. Results demonstrated TNF-α-induced nuclear translocation of NF-κB, while treatment with AKE at 50 or 100 μg/mL inhibited such nuclear translocation (Fig. 6B). The NF-κB pathway plays a crucial role in triggering the activation of HUVEC in response to TNF-α stimulation (Monaco and Paleolog, 2004). In the absence of stimulation, NF-κB resides in the cytoplasm in association with IκB proteins. Upon inflammatory stimuli, including TNF-α, IκBα undergoes phosphorylation and degradation, allowing NF-κB to translocate to the nucleus, initiating the transcription of various genes, including E-selectin, ICAM-1, and VCAM-1 and (Collins, 1993; Lu et al., 2012). In conclusion, our study suggests that AKE’s anti-inflammatory effects in TNF-α-stimulated HUVECs are primarily mediated through the selective inhibition of the JNK/NF-κB signaling pathway, offering potential therapeutic implications for vascular inflammation.
4. Conclusions
This study aimed to evaluate the antioxidant, cytotoxic, and anti-inflammatory properties of AKE in HUVECs treated with TNF-α. DPPH and ABTS assays determined the radical scavenging activities of AKE, yielding IC50 values of 375.24 ±8.56 μg/mL and 568.12±9.23 μg/mL, respectively. WST assay revealed no cytotoxicity of AKE up to 200 μg/mL. Flow cytometric analysis showed that AKE significantly reduced intracellular ROS levels in TNF-α-treated HUVECs, with enhanced activities of antioxidant enzymes such as SOD, GSH, and catalase. Western blot analysis demonstrated that AKE inhibited the expression of vascular adhesion molecules (E-selectin, ICAM-1, and VCAM-1) and selectively reduced the phosphorylation of JNK without affecting ERK and p38 in the MAPK pathway. Additionally, AKE decreased the phosphorylation of IκBα and NF-κB, inhibiting NF-κB nuclear translocation in TNF-α-stimulated HUVECs. The observed vascular endothelial cells protection effect of AKE may be attributed to its flavonoid content, which includes chalcone, xanthoangelol, isobavachalcone, luteolin, and cynaroside (Kil et al., 2017). These flavonoids are known for their potent antioxidant and anti-inflammatory properties (Zhang et al., 2011) which likely contribute to the overall beneficial effects of AKE in mitigating oxidative stress and inflammation in vascular cells. Further research into these specific flavonoids could provide deeper insights into their individual and synergistic roles in the protective effects of AKE. These findings suggest that AKE possesses significant antioxidant and anti-inflammatory properties, making it a promising candidate for managing oxidative stress and inflammatory vascular conditions.